Most antipsychotics (with the exception of Xanomeline and maybe Clozapine) operate in the so-called “Dopamine hypothesis” in which altered dopaminergic functioning in the brain leads to the different symptoms. Hyperdopaminergia in the mesolimbic (or corticostriatal) pathway and hypodopaminergia in the corticolimbic pathway. The source of this dysregulation is not entirely understood.1, 2
Typical antipsychotics (TAs)
TAs are mainly antagonists for the D2 receptor, a postsynaptic dopamine receptor. By antagonizing the postsynaptic receptor we can treat positive symptoms associated with hyperdopaminergia.2
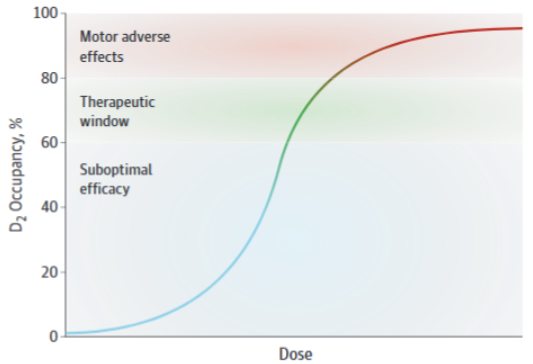
These agents are known as “typical” for their propensity to induce motor AEs like drug induced parkinsonism and tardive dyskinesia. When looking at the neuroanatomy, we see that in order to regulate fine motor movement, the dorsal striatum uses dopamine, and excessive blockade of D2 receptors leads to dysregulation of dopamine signalling in the dorsal striatum, leading to these symptoms. It is said that blocking >80% of D2 receptors leads to very high rates of motor AEs (MAEs) due to impaired dorsal striatum functioning. But blocking at least 65% of D2 receptors is necessary for antipsychotic activity. Blocking 65-80% of D2 receptors is considered the “sweet-spot” for therapeutic results.1,2 Albeit with very heterogeneous responses and less than good correlation.1
When I was learning about antipsychotics, I always wondered how blocking D2, an inhibitory metabotropic receptor (and a presynaptic autoreceptor), reduced “limbic hyperdopaminergia”, in my mind, blocking D2 would lead to even more arousal! In truth, I don’t think we actually know how they work, but there are a lot of ideas going on:
First there is the concept of dopamine depolarization block, obtained from healthy animal models. After 3-4 weeks of antipsychotic administration, an inhibition of dopamine neuron firing was blocked. An interesting finding was that for TAs, this depolarization block occurred in both the mesolimbic system and the nigrostriatal system (therefore producing motor effects), and atypical antipsychotics would only cause depolarization block in the mesolimbic system.3
The main issue with this is that it does not represent the onset of action of antipsychotics, you don’t need to administer them repeatedly for 3+ weeks to achieve symptom remission. The author of the paper cited also talks about a lack of evidence for DA hyperactivity.3 According to animal models of schizophrenia, a dysregulated DA system (aka a hyperresponsive one) is more responsive to antipsychotics, leading to depolarization block in a shorter time frame (days instead of weeks). This also reflects the clinical observation that more psychotic patients respond faster to antipsychotics.3, 4
Basically: blocking D2 produces an overactivation of DA neurons and this induces depolarization. If the DA neurons were previously overactivated like in schizophrenic patients (presumably), this blockade would happen more quickly.
D2 antagonism may also lead to interactions with beta-arrestin 2 (BA2) signalling. Beta-arrestins in general serve to sterically hinder G-coupled proteins, and are one of the mechanisms behind receptor desensitization, but at least for BA2, they can serve as multiprotein scaffold (proteins get recruited by BA2 to modulate their behavior), and like this do cellular signalling on their own.5 There are also some older (2007) papers6 that talk about D2 mediated regulation of AKT signalling.
The D2 receptor is a G-coupled protein that, when agonized, leads to inhibition of adenylyl cyclase, this leads to further cascading effects, this is the G protein dependent signalling (Figure 2).7 On the other hand, it is known that agonism of the D2 receptor also leads to inhibition of AKT. This happens through the recruitment of beta-arrestin, which forms complexes with PP2A and AKT. PP2A inhibits AKT, so the complex formed leads to inhibition of AKT. One of the roles of AKT is to inhibit GSK3, so by inhibiting AKT we get more GSK3 activity.7
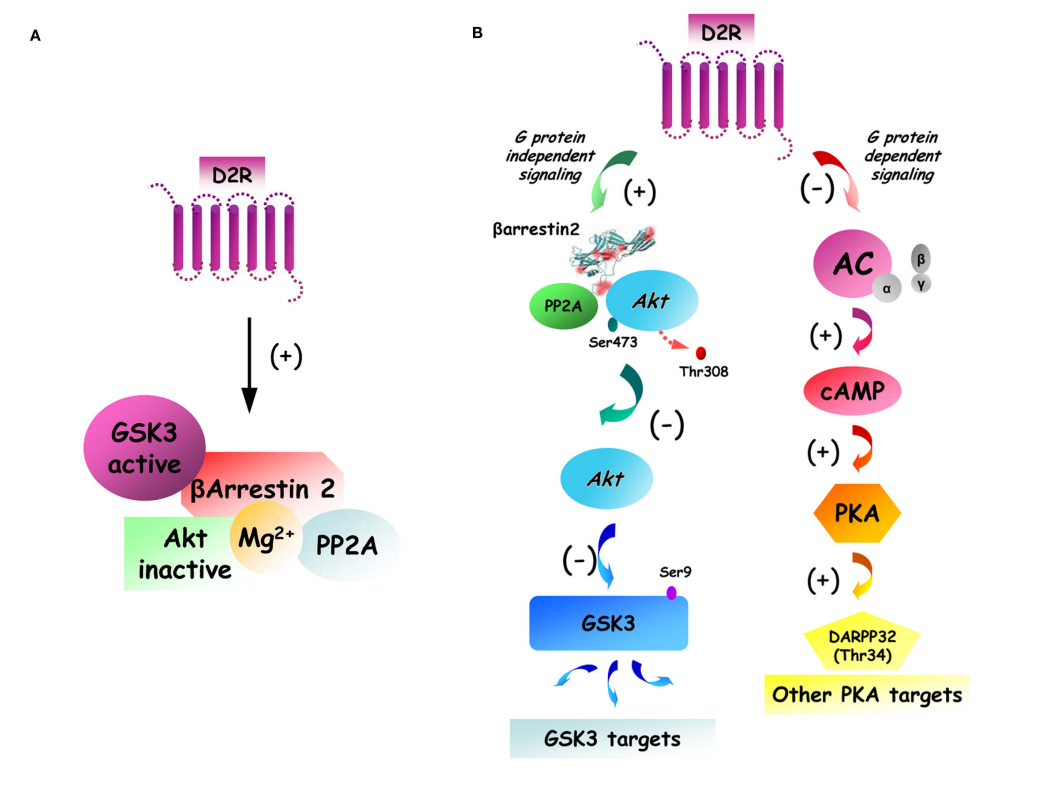
To sum up the biochemistry: we have the “normal” G protein-dependent signalling, then we have the recruitment of beta arrestins to inhibit AKT and reduce GSK3 inhibition. You can probably imagine that D2 antagonism would lead to less AKT inhibition and more GSK3 inhibition.
Atypical antipsychotics/Second generation antipsychotics
Atypical antipsychotics can have multiple mechanisms, but most of them still act on dopamine receptors.
Some of them are partial agonists for the D2 receptor, by having a low intrinsic effect on dopamine receptors, you can avoid most of the MAEs, in fact, this is exactly why these antipsychotics are called “Atypical”, because they don’t produce MAEs like typical antipsychotics do.2
Some atypical antipsychotics also act as 5-HT2a antagonists, weak partial agonists or inverse agonists. These receptors play a role in the regulation of glutamatergic and dopaminergic neurons. Clinically, it has a big impact in motor symptomatology.1, 2
In the motor striatum, 5-HT2a activity indirectly causes an inhibition of dopamine release. The specific mechanism goes as follows: 5-HT2a stimulates glutamatergic neurons which in turn stimulate gabaergic neurons (inhibitory neurons) which inhibit dopamine release in the motor striatum. Blocking this receptor would allow a disinhibition of dopamine release restoring functionality, hence the lower rate of MAEs (Figure 3)2
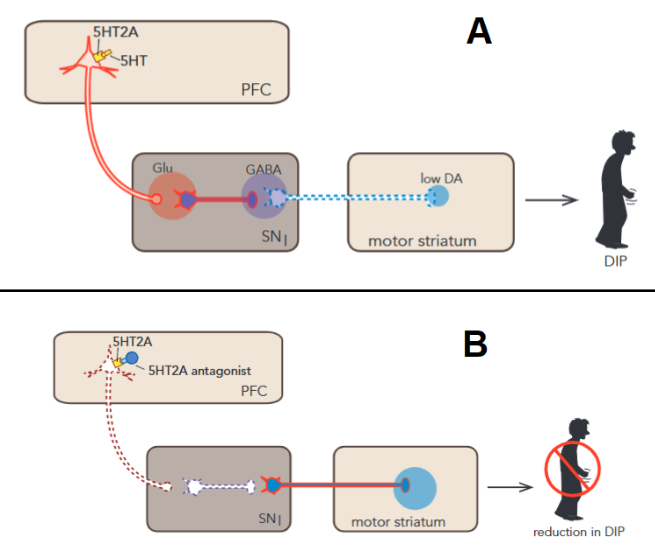
BTW I cannot end this article without mentioning that there are many hypotheses about the possible role of 5HT2A in the antipsychotic effect of drugs like Clozapine
-
References
- Jonathan M. Meyer, “How antipsychotics work in schizophrenia: a primer on mechanisms”, 2024. DOI: 10.1017/S1092852924002244
- S. Stahl, “Stahl's Essential Psychopharmacology: Neuroscientific Basis and Practical Applications”, 5th ed, 2021.
- A. Grace, D. Uliana, “Insights into the Mechanism of Action of Antipsychotic Drugs Derived from Animal Models: Standard of Care versus Novel Targets”, 2023. DOI: 10.3390/ijms241512374
- S. Miyamoto, N. Miyake, L. Jarskog, “Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents”, 2012. DOI: 10.1038/mp.2012.47
- J.M. Beaulieu, T. Sotnikova, et al, "An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior", 2005. DOI: 10.1016/j.cell.2005.05.012
- J.M. Beaulieu, T. Sotnikova, et al, “Regulation of Akt Signaling by D2 and D3 Dopamine Receptors In Vivo”, 2007. DOI: 10.1523/JNEUROSCI.5074-06.2007
- J.M. Beaulieu, T. Del’Guidice, et al, “Beyond cAMP: the regulation of Akt and GSK3 by dopamine receptors”, 2011. DOI: 10.3389/fnmol.2011.00038