Back in the day, when you started screaming out in public about your brand new OS dedicated entirely to the worship of God, people in white robes would come and give you a really comfortable shirt that you could not take off, at least until you got put into the pillow room. (figure 1) Afterwards, you would be treated by cutting edge techniques such as Medically Induced Hypoglycemic Coma and Electroshocks. If you were really lucky, you would get the nobel-prize winning surgical procedure known as lobotomy.

Fortunately enough, advances in anti-allergy research led to a wonderful compound called Chlorpromazine1,2, this drug was discovered to have calming properties by the french surgeon Henri-Marie Laborit, who was seeking to enhance the effects of anesthetics with various chemical agents. This led to psychiatrists Jean Delay and Pierre Deniker to try out this drug on psychiatric inpatients at St. Anne’s Hospital, soon discovering its great tranquilizing effects in psychotic and agitated patients.1,2
Antipsychotics would then allow countless psychiatric patients to enjoy relatively stable lives, that is, if they could withstand the often severe adverse events (AEs) these drugs inflicted. A particularly common and pervasive side-effect caused by these early antipsychotics are motor symptoms3, (known previously as extrapyramidal symptoms) which are essentially pharmacologically-induced parkinson's disease. This led to the development of newer antipsychotics that try to reduce this safety issue. Even with these newer generation agents, there are many safety and efficacy issues surrounding antipsychotic therapy.
A major problem behind the lack of efficacy for antipsychotics is the fact that most of them act on the same pharmacological target, the D2 dopamine receptor. Most antipsychotics target the D2 receptor, with the only exceptions being Clozapine (which still antagonizes D2 but weakly) and Xanomeline3, the latter being the focus of these articles. In order to understand how this agent known as KarXT (combination of Xanomeline and Trospium chloride) works, we must first talk about schizophrenia and the current pharmacological treatments for this psychiatric disorder.
Schizophrenia
This chronic psychiatric condition is prevalent in slightly less than 1% of the population1 and is characterized by a large variety of symptoms sometimes defined as three distinct clusters: positive symptoms which include disordered thoughts and hallucinations (among other symptoms). Most people suffering from schizophrenia seek professional advice only after their first psychotic episode, which is essentially the maximum expression of positive symptomatology; Negative symptoms and cognitive symptoms1, which although important for the quality of life of the patient, are often-times neglected or not mentioned when it comes to discussion about this condition. The mechanisms and neurobiological features of negative and cognitive symptoms are very murky so I wont focus on them too much.
Positive Symptoms
By far the most recognizable part of schizophrenia. Hallucinations, disordered thinking, erratic and/or odd behaviour, etc. This is the main cluster that current pharmacological therapy is able to treat. Unfortunately, antipsychotic therapy suffers from a high rate of treatment resistance with around 30% of patients not responding to first generation antipsychotics and about 50% of them responding only partially.3, 1 For patients that are treatment resistant (TRS), the only approved treatment is Clozapine. And even with this, the response rate with this drug is only about 30%.1
There are many different hypotheses that attempt to explain the pathophysiology behind positive symptomatology, the most successful one being the dopamine hypothesis. This hypothesis postulates that dysfunction of dopamine signalling (both hyperdopaminergia and hypodopaminergia) in the brain leads to the pathogenesis of schizophrenia.3, 4 This hypothesis was developed after neuropharmacologist Arvid Carlsson connected the loss of dopamine neurons in Parkinson’s disease, and antipsychotic medication causing a form of parkinsonism. This made him postulate that antipsychotics must be blocking dopamine, and that excessive dopamine signalling causes psychotic episodes. This discovery garnered him a nobel prize!3
When it comes to the neurobiology of the dopamine hypothesis, a more classical look (some authors even refer to this as dogma)5 postulates a mesolimbic hyperdopaminergia. The mesolimbic pathway (Figure 2) consists of dopaminergic neurons originating from the ventral tegmental area (VTA) to the ventral striatum. This pathway is associated with reward and behavior. Overstimulation of this pathway (according to this so-called dogma) is associated with psychotic episodes and positive symptomatology.5
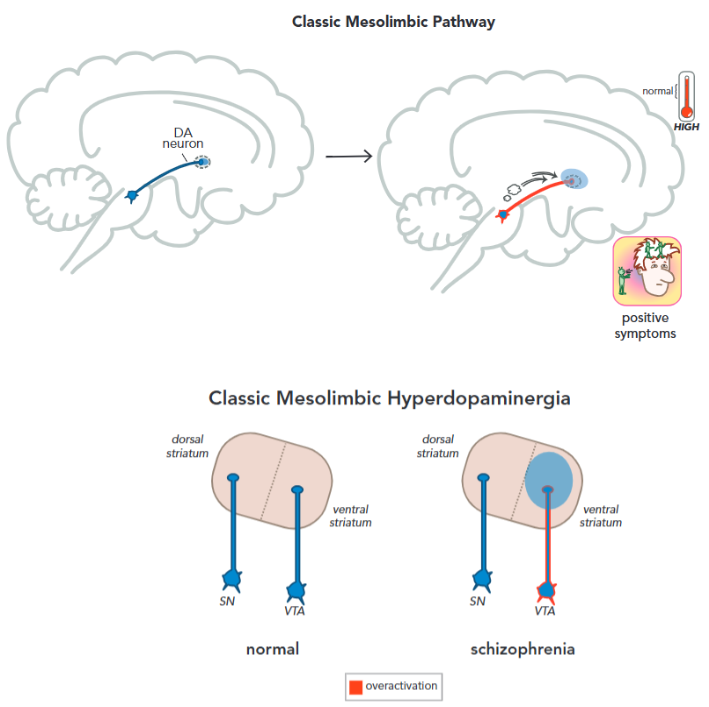
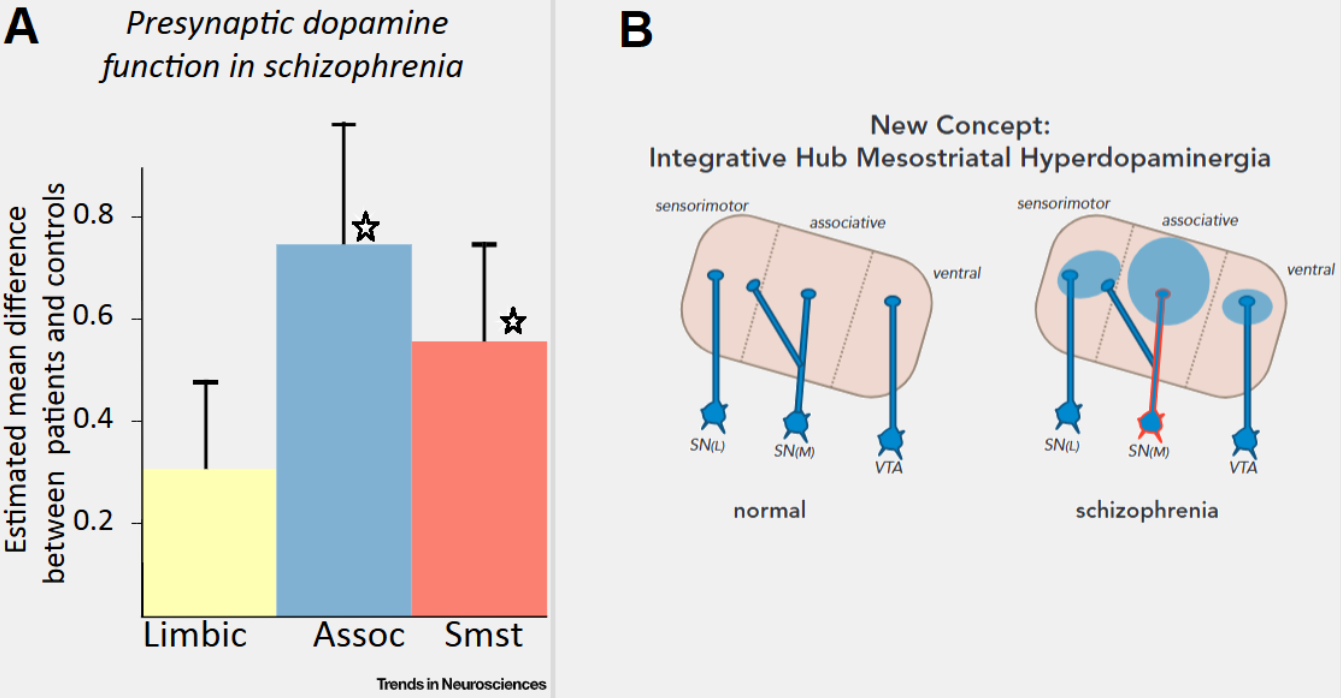
Newer studies4, 5 indicate that mesolimbic hyperdopaminergia may not come just from the VTA as originally thought, some scans on unmedicated patients with schizophrenia showed a greater overactivation of the associative striatum (dorsal striatum) alongside the limbic striatum (ventral striatum). The associative striatum is not regulated by the VTA, but the medial substantia nigra. A meta-analysis (McCutcheon, R. et al. 2019,5) of PET scans also concluded that dopaminergic dysfunction is greatest in the associative striatum.4, 5 (Figure 3) Previous analysis would struggle to differentiate between the different parts of the striatum, defining only two parts: dorsal (movement related) and ventral striatum (emotion related); We now know that in reality, the “emotional component” of the striatum isn't necessarily limited to the ventral striatum, but also distributed between the associative and dorsal striatum.4
Beyond the dopamine hypothesis of schizophrenia, there is a lot of research being done on the glutamate hypothesis. This hypothesis was proposed after it was noted that NMDA antagonists (dissociatives like Ketamine and PCP) induce temporary schizophrenia-like symptoms (positive, negative, and cognitive symptomatology) and that psychosis induced by ketamine is clinically difficult to distinguish from primary psychosis of schizophrenia. There is also a genetic factor that yields credit to the glutamate hypothesis, about 30% of all risk genes for schizophrenia encode glutamate transmission related proteins. Another interesting data point is the presence of anti-NMDAR autoantibodies in about 5% of patients with schizophrenia. It is thus postulated that a dysfunction in the NMDAR receptor and abnormalities in glutamate transmission.6, 7 Something remarkable about agents like Iclepertin (Glycine transporter inhibitor) and D-cycloserine (NMDAR partial agonist) tested for schizophrenia (as an adjunct to antipsychotic therapy) is the supposed reduction of negative and cognitive symptomatology.6
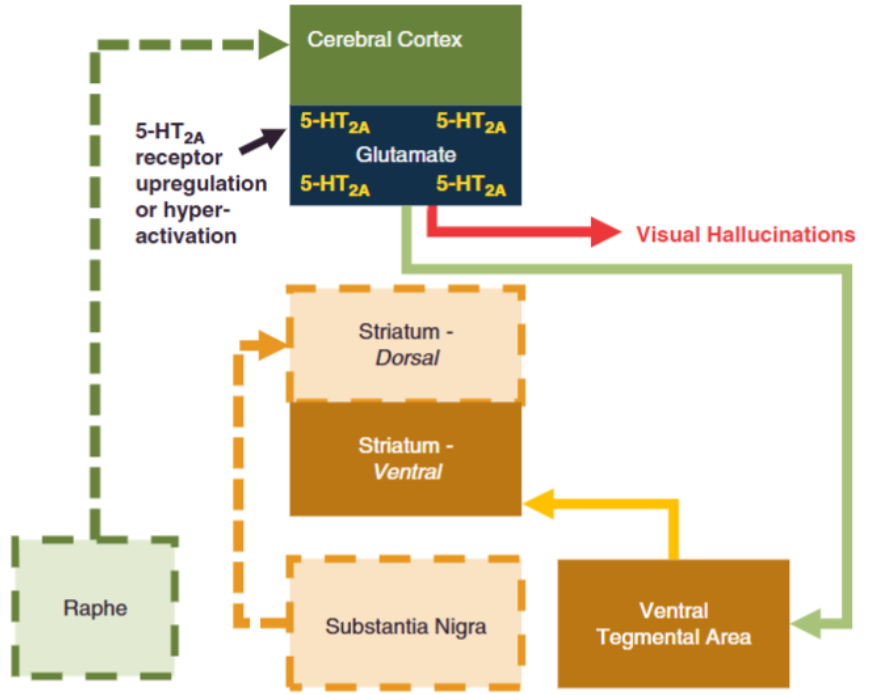
There are many other hypotheses behind the pathophysiology of schizophrenia, some of them are more accepted or integrated with other mechanism such as the serotonin hypothesis (figure 4), and other mechanisms that sound more obscure such as the Phospholipid hypothesis8 and an exogenous cannabinoid hypothesis9 I shall discuss them in some other occasion, maybe.
Negative Symptoms
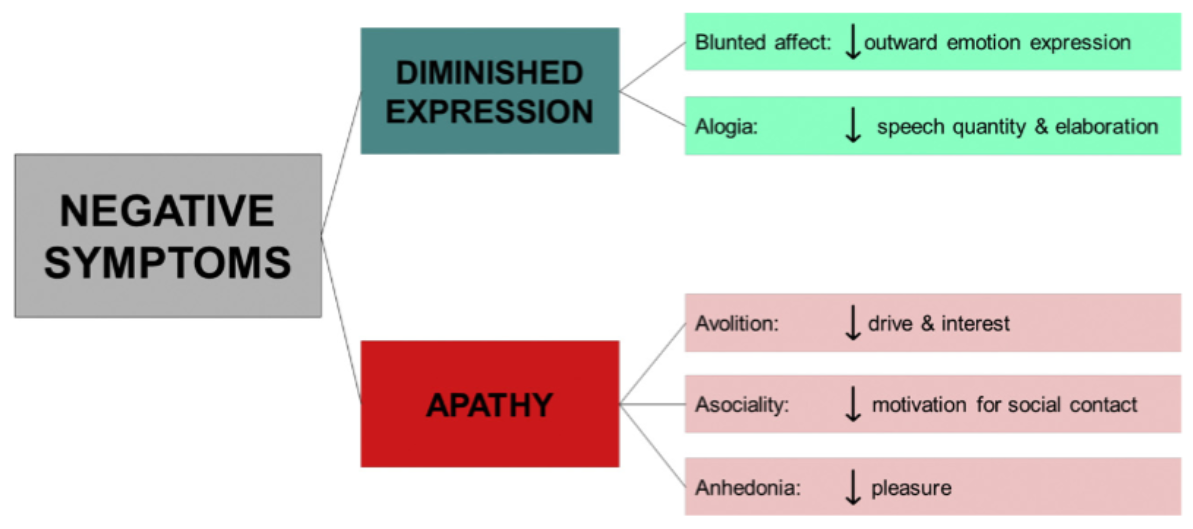
There are five major factors behind this cluster: Anhedonia (inability to feel pleasure), avolition (apathy), social withdrawal, alogia (akin to speech disorders), emotional flattening and blunting.11 What you may notice right away is that some of these symptoms can be easily confused with AEs to antipsychotic therapy, (e.g motor symptoms may cause emotional flattening and amimia or diminished facial expression) or affective disorders (e.g depression), these are known as secondary negative symptoms. This makes differential diagnoses necessary in order to properly identify primary negative symptomatology from secondary symptomatology.11
Unfortunately, we know little about the aetiology of these symptoms and as of 2025, there is not a single approved drug for the treatment of negative symptoms in schizophrenic patients and unlike positive symptoms, they do not tend to resolve spontaneously. It is a major unmet medical need.12
Negative symptomatology usually begins in the prodrome (before the disease manifests, “prepsychotic”) stage and may present as diminished social activity and lack of energy, and can develop into dementia, and in the worst cases, marasmus, a condition so severe I will simply quote Sergey N. Mosolov and Polina A. Yaltonskaya (2022)11:
“Marasmus - fragmentation of the personality to such an extent that the individual no longer presents a unified, predictable set of beliefs, attitudes, traits, and behavioral responses; profound dementia with loss of contact with the environment and complete disappearance of interests and desires. The food and sexual instincts are preserved, but severe physical exhaustion, trophic changes in the skin, dystrophy of the internal organs, increased bone fragility could be observed. Marasmus with a full disintegration of the personality is the most severe type of negative syndrome.”
When it comes to the neurobiology of negative symptomatology, there are multiple different components that are thought to originate them, for instance, the dopamine hypothesis of schizophrenia postulates that its the result of hypoactivity in the mesocortical dopamine pathway4 (Figure 6), while some other authors implicate hypodopaminergic activity in the mesocortical pathways, and the dorsal striatum. Some also make the point that, instead of discrete regions of the brain being dysfunctional (like the limbic system), irregularities in distributed networks like the frontocortico-temporal and cortico-striatal networks lead to the emergence of negative symptomatology.14
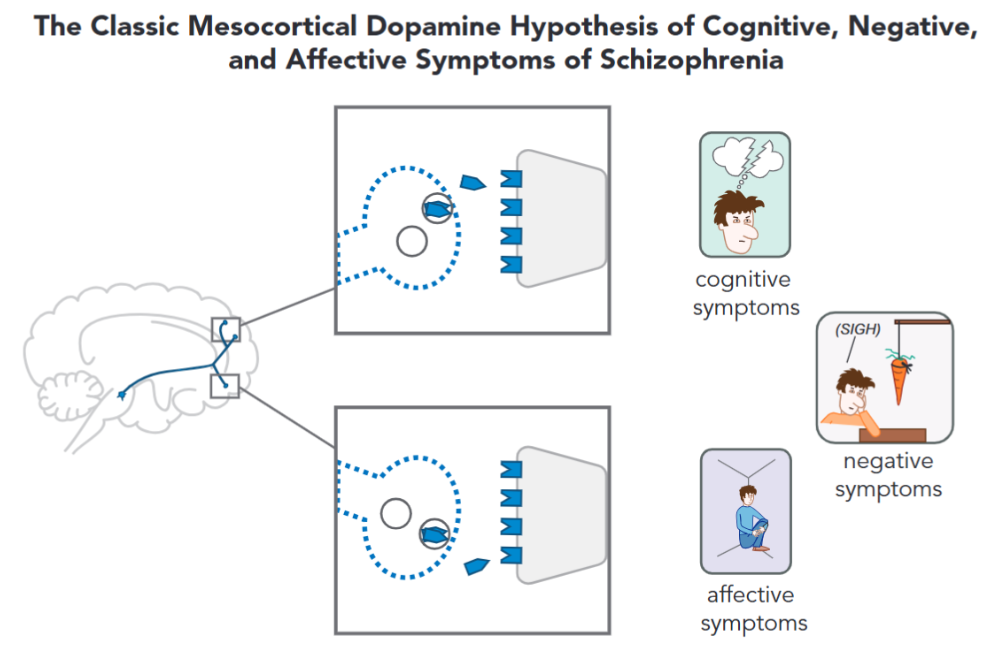
Cognitive Symptoms
These symptoms refer to the deterioration in cognitive capabilities, such as an inability to organize one’s life, a deficit of semantic, explicit, and working memory, attention deficits. These symptoms, like negative ones, do not improve sporadically over time, are largely resistant to treatment, have no approved drugs that attempt to treat it, and tend to get worse. Generally, the severity of cognitive symptoms are a great predictor of the long-term prognosis, even greater than positive symptoms.16
The neurobiology of these symptoms is not clear at all, but some authors mention hypoactivity in the mesocortical pathway to the dorsolateral prefrontal cortex. Disregulations in the prefrontal cortex are thought to directly hinder cognitive and executive functioning.4, 16 Other mechanisms behind the cognitive symptoms include NMDA dysfunction (glutamate hypothesis), aberrant hippocampal activity, and hippocampal-prefrontal communication.16
Of note, is the possibility that antipsychotic medication produces cognitive deterioration by itself, on top of the cognitive decline associated with schizophrenia. For example, Clozapine and quetiapine are strongly anti cholinergic and anticholinergic burden is implicated with cognitive decline. And beyond antipsychotics, patients with schizophrenia usually have polypharmacy which also contributes to anticholinergic burden.17
-
References
- S. Miyamoto, N. Miyake, L. Jarskog, et al, “Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents”, 2012. DOI: 10.1038/mp.2012.47
- F. López-Muñoz, C. Alamo, et al, “History of the discovery and clinical introduction of chlorpromazine”, 2005. DOI: 10.1080/10401230591002002
- Jonathan M. Meyer, “How antipsychotics work in schizophrenia: a primer on mechanisms”, 2024. DOI: 10.1017/S1092852924002244
- S. Stahl, “Stahl's Essential Psychopharmacology: Neuroscientific Basis and Practical Applications”, 5th ed, 2021.
- R. McCutcheon, A. Abi-Dargham, D. Howes, “Schizophrenia, Dopamine and the Striatum: From Biology to Symptoms”, 2019. DOI: 10.1016/j.tins.2018.12.004
- Yota Uno, J. Coyle, “Glutamate Hypothesis in Schizophrenia. Psychiatry and Clinical Neurosciences”, 2019. DOI: 10.1111/pcn.12823
- R. Okuba, M. Okada, E. Motomura, “Dysfunction of the NMDA Receptor in the Pathophysiology of Schizophrenia and/or the Pathomechanisms of Treatment-Resistant Schizophrenia”, 2024. DOI: 10.3390/biom14091128
- D. Horrobin, C. Bennett, "The Membrane Phospholipid Concept of Schizophrenia", 1999. DOI: 10.1007/978-3-642-47076-9_19
- R. Little, D. D’Mello. “A Cannabinoid Hypothesis of Schizophrenia: Pathways to Psychosis”, 2022. PMID: 36204167
- S. Stahl, “Beyond the dopamine hypothesis of schizophrenia to three neural networks of psychosis: dopamine, serotonin, and glutamate”, 2018. DOI: 10.1017/S1092852918001013
- S. Mosolov, P. Yaltonskaya, “Primary and Secondary Negative Symptoms in Schizophrenia”, 2022. DOI: 10.3389/fpsyt.2021.766692
- J. Kantrowitz, C. Correll, et al, “New Developments in the Treatment of Schizophrenia: An Expert Roundtable”, 2023. DOI: 10.1093/ijnp/pyad011
- Vikaas Sohal, "Neurobiology of schizophrenia", 2024. DOI: 10.1016/j.conb.2023.102820
- C. Correll, N. Schooler, “Negative Symptoms in Schizophrenia: A Review and Clinical Guide for Recognition, Assessment, and Treatment. Neuropsychiatric Disease and Treatment”, 2020. DOI: 10.2147/ndt.s225643
- Lina Zeldovich, “Cold parenting? Childhood schizophrenia? How the diagnosis of autism has evolved over time”, 2018. Obtained from: https://www.science.org/content/article/cold-parenting-childhood-schizophrenia-how-diagnosis-autism-has-evolved-over-time
- E. Simpson, C. Kellendonk, E. Kandel, “A Possible Role for the Striatum in the Pathogenesis of the Cognitive Symptoms of Schizophrenia”, 2010. DOI: 10.1016/j.neuron.2010.02.014
- Y. Joshi, M. Thomas, et al, “Anticholinergic Medication Burden–Associated Cognitive Impairment in Schizophrenia”, 2021. DOI: 10.1176/appi.ajp.2020.20081212